Genomic language models (GLMs) are multiplying fast and differ widely in architecture, training
paradigm, and data. Comparing them in a principled way matters for building better models and for
picking the right one. Yet today this relies on downstream benchmarks, which are
phenotype-like: they record how a model behaves on a task, not how models relate
to one another.
The same gap appears in population genetics (Fig. 1a, b): benchmark scores
act as a phenotype matrix, but the genotype-derived marker matrix that reveals population
structure has no counterpart for GLMs. A model's internal constitution (architecture,
weights, tokenizer, objective, data) is non-aligned across models and cannot be compared directly.
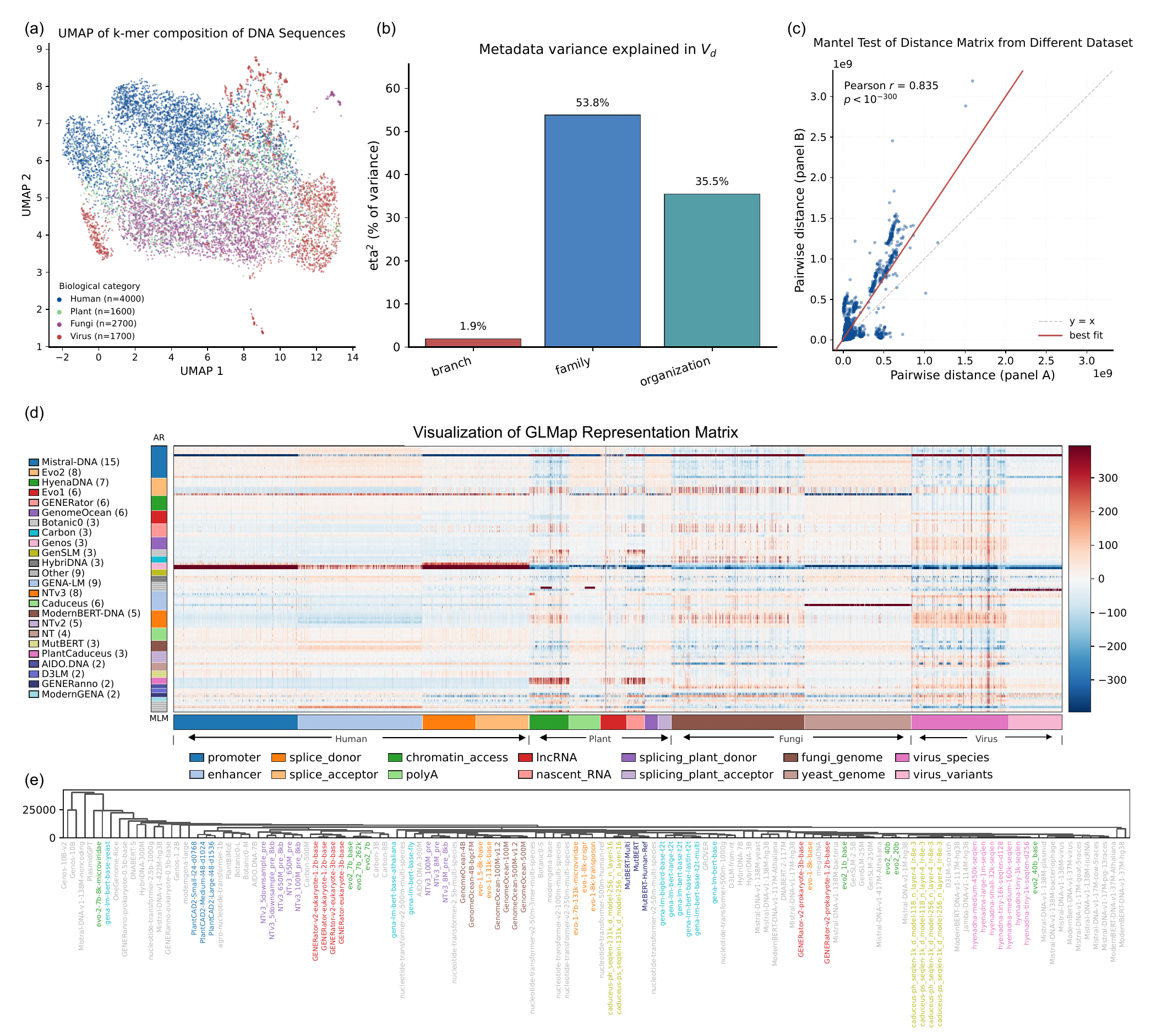
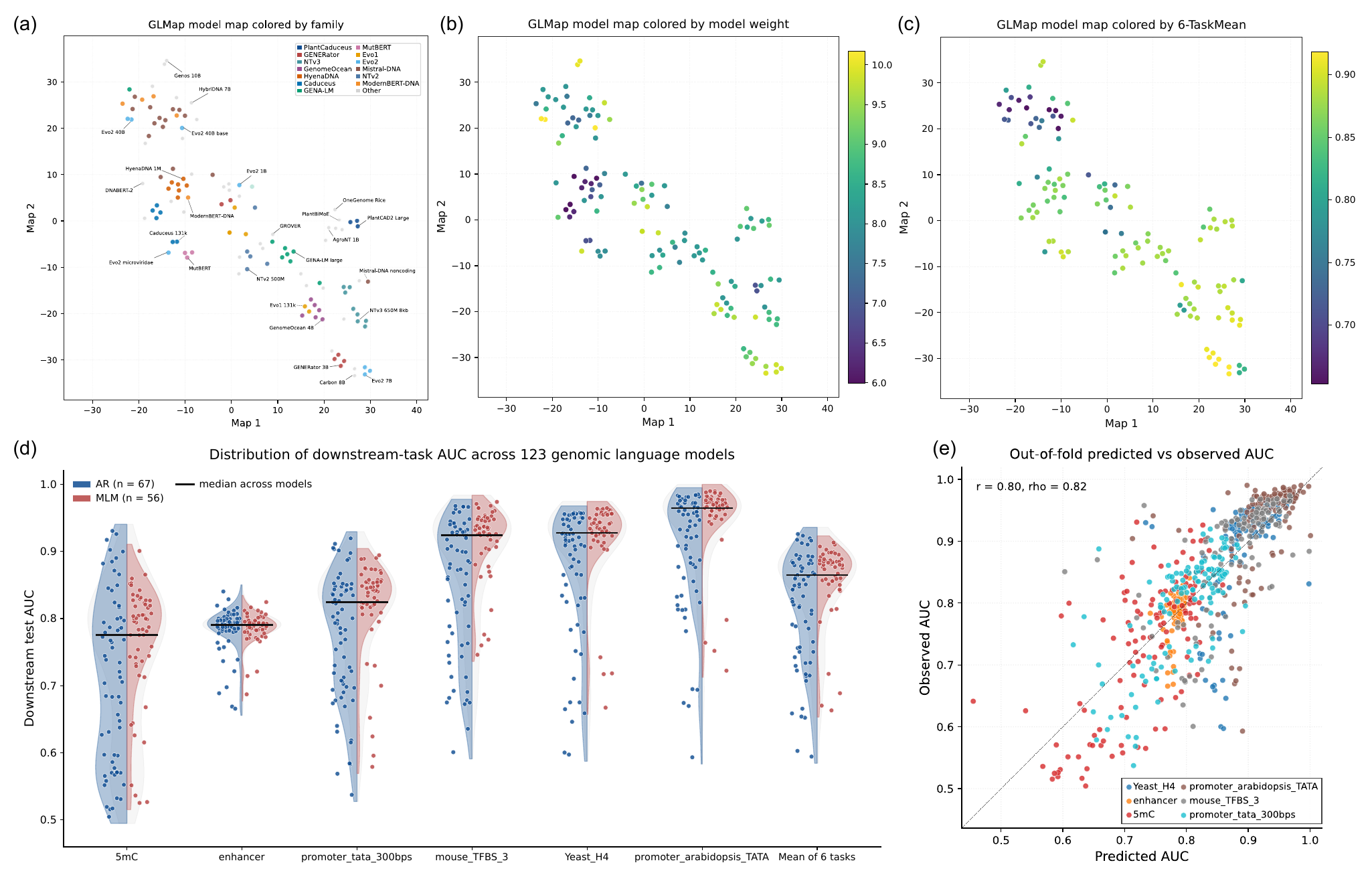
We propose GLMap, a standardized, model-agnostic framework (Fig. 1c): we score a
fixed panel of DNA probes with every frozen model and record its (pseudo-)log-likelihood responses,
assembling a functional marker matrix over models and probes.
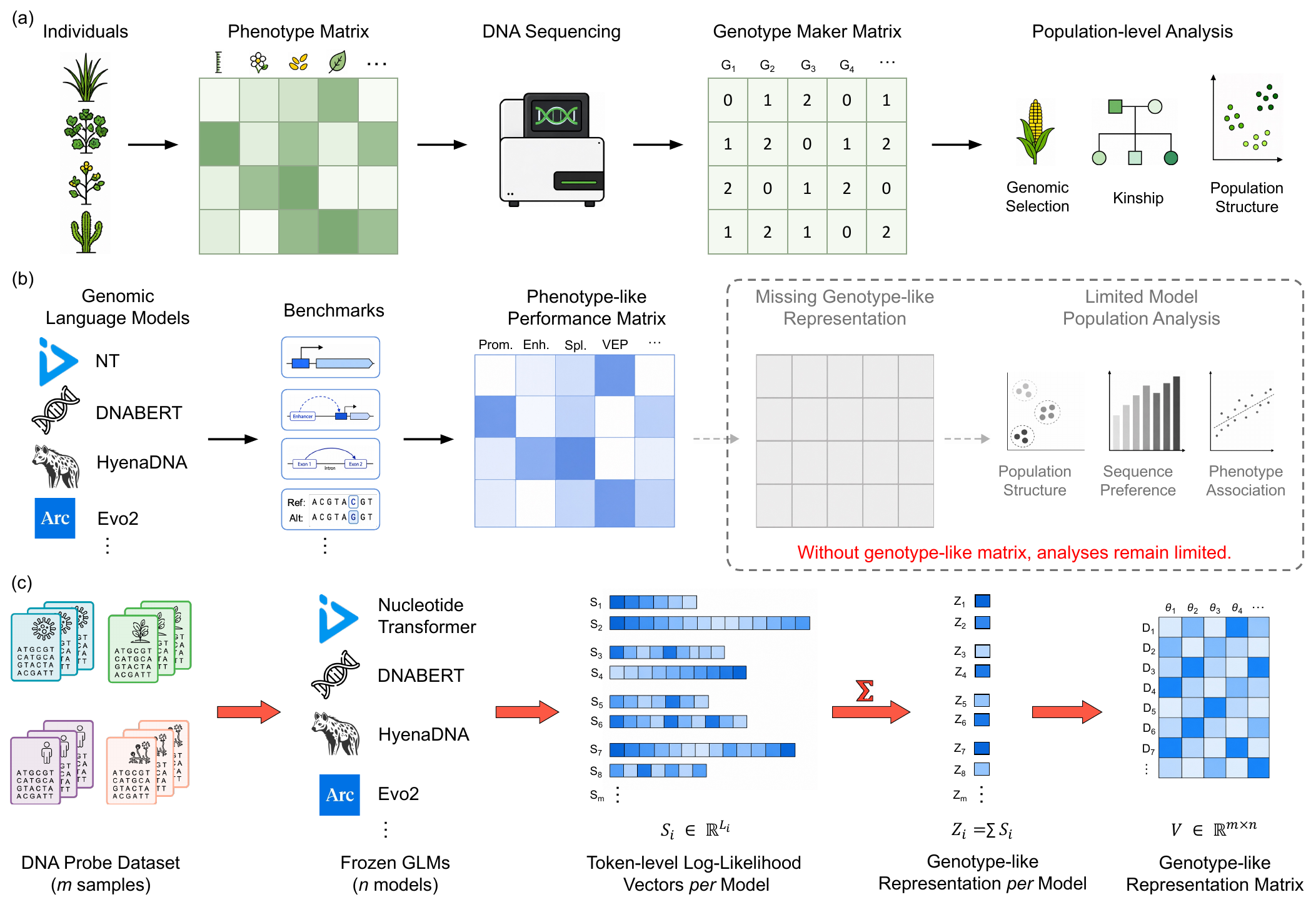
Fig. 1.
GLMap probes every genomic language model with the same fixed panel of DNA sequences and records
its likelihood responses, producing a functional marker matrix over models and probes, a
task-independent way to place heterogeneous models in a common space.